PyGenePlexus
PyGenePlexus is a Python package for running the [GenePlexus] model.
PyGenePlexus enables researchers to predict novel genes similar to their genes of interest based on their patterns of connectivity in genome-scale molecular interaction networks.
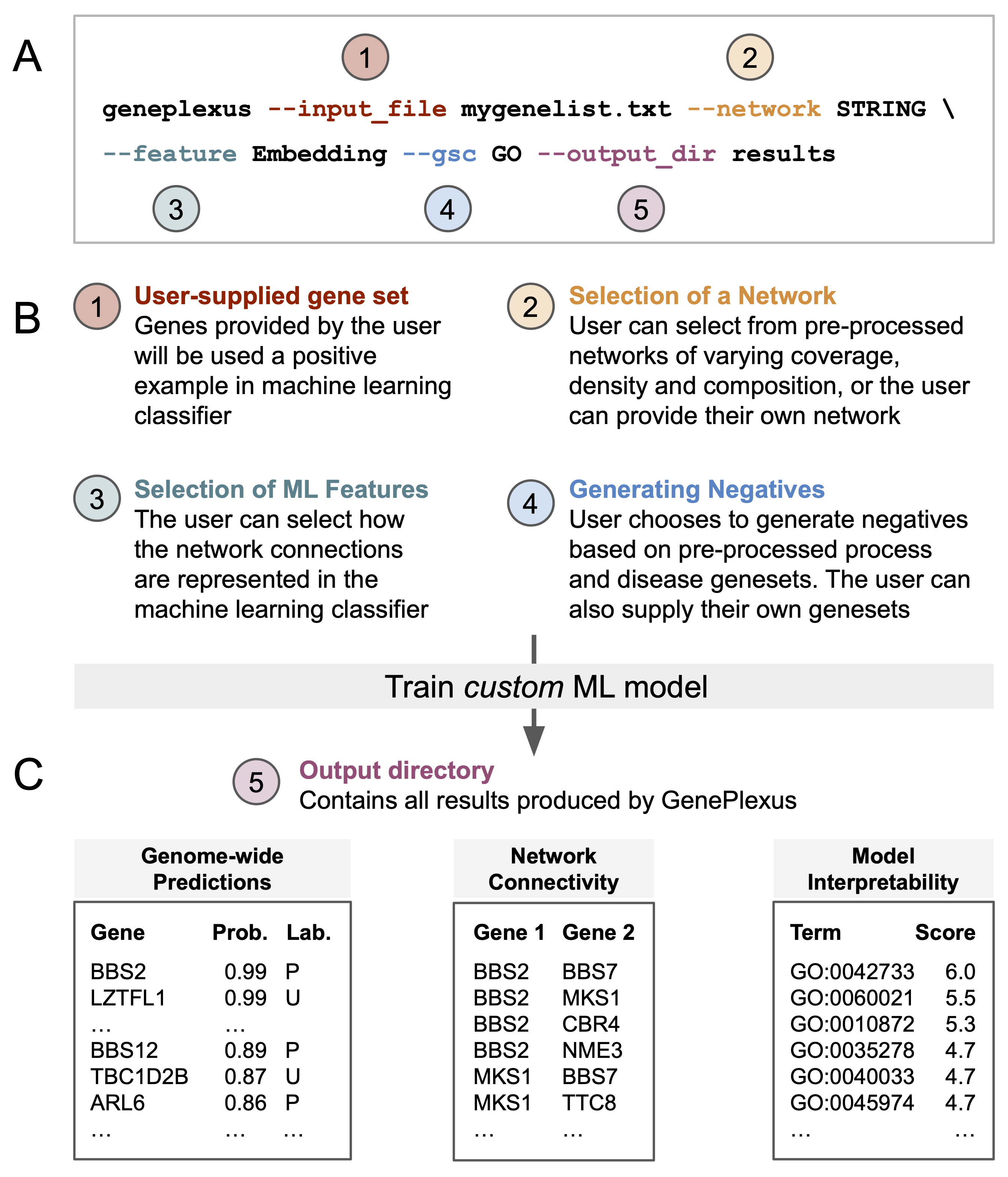
Overview of PyGenePlexus
Given a list of input genes and a geneset collection (GSC) to help select negative examples, the package trains a logistic regression model using one of three network derived features (adjacency, influence, or embedding) and generates the following outputs
Genome-wide prediction of how functionally similar a gene is to the input gene list. Evaluation of the model is provided by performing k-fold cross validation. The default is 3-fold cross validation when a minimum of 15 input genes are supplied. These parameters can be changed when accessing the Python class. PyGenePlexus does not enforce a minimum or maximum number of genes, and we note evaluations of the model were carried out for gene sets ranging between 5 and 500 genes. See
fit_and_predict()(Optional) Interpretability of the model is provided by comparing the model trained on the user gene set to models pretrained on 1000’s of known gene sets from [GO] bioloigcal proceses and [DisGeNet] diseases. See
make_sim_dfs()(Optional) Interpretability of the top predicted genes is provided by returning their network connectivity.
make_small_edgelist()
Note
Links to other GenePlexus products
Quick start
PyGenePlexus comes with an easy to use command line interface (CLI) to run the full GenePlexus pipeline given an input gene list. Go get started, install via pip and run a quick example as follows.
pip install geneplexus
geneplexus -i my_gene_list.txt --output_dir my_result
Note that you need to supply the my_gene_list.txt file, which is a line
separated gene list text file (NCBI Entrez IDs, Symbol or Ensembl IDs are
accepted). An example can be found on the
GitHub page under
example/input_genes.txt. More info can be found in PyGenePlexus CLI.
Warning
All necessary files for a specific selection of parameters (network,
feature, and gene set collection) will be downloaded automatically and
saved under ~/.data/geneplexus. User can also specify the location of
data to be saved using the --output_dir argument. The example
provided will download files that occupy ~300MB of space.
Using the API
A quick example of generating predictions using an input gene list. More info can be found in PyGenePlexus API.
>>> from geneplexus import GenePlexus
>>> input_genes = ["ARL6", "BBS1", "BBS10", "BBS12", "BBS2", "BBS4",
... "BBS5", "BBS7", "BBS9", "CCDC28B", "CEP290", "KIF7",
... "MKKS", "MKS1", "TRIM32", "TTC8", "WDPCP"]
>>> gp = GenePlexus(net_type="STRING", features="Embedding", gsc="DisGeNet",
... input_genes=input_genes, auto_download=True, log_level="INFO")
>>> df_probs = gp.fit_and_predict()[1]
>>> df_probs.iloc[:10]
Entrez Symbol Name Probability Known/Novel Class-Label Rank
0 8100 IFT88 intraflagellar transport 88 0.995984 Novel U 1
1 585 BBS4 Bardet-Biedl syndrome 4 0.992909 Known P 2
2 261734 NPHP4 nephrocystin 4 0.990705 Novel U 3
3 91147 TMEM67 transmembrane protein 67 0.986072 Novel U 4
4 9657 IQCB1 IQ motif containing B1 0.983366 Novel U 5
5 582 BBS1 Bardet-Biedl syndrome 1 0.979287 Known P 6
6 200894 ARL13B ADP ribosylation factor like GTPase 13B 0.977565 Novel U 7
7 8481 OFD1 OFD1 centriole and centriolar satellite protein 0.974288 Novel U 8
8 80184 CEP290 centrosomal protein 290 0.963544 Known P 9
9 54903 MKS1 MKS transition zone complex subunit 1 0.960611 Known P 10
Supported networks
Currently, GenePlexus come with four networks, including [BioGRID], [STRING] (default), [STRING-EXP], and [GIANT-TN]. Prediction using a custom network can also be done, see Using custom networks. However, when using a custom network, the model similarity analysis cannot be done due to the lack to pretrained models.
Using PyGenePlexus
Package reference
Medthods and guidelines
Appendix