Guidelines for Selecting Parameters
The first step is choosing the network. If your gene set of interest comes from a curated database or it was generated while studying a specific process/pathway or disease, the best network to choose is STRING as this network is a highly curated network that uses prior knowledge from gene set databases in building the network. If you would like to only consider experimental interactions, then the network to use is STRING-EXP, and if you would further only like to consider physical interactions, choose BioGRID. The GIANT-TN network is best to use for two cases. First, since it offers the highest gene coverage, it enables the user to see predictions on many more understudied genes. Second, as GIANT-TN is a very dense network that does not directly incorporate gene set database information, this network performs well on larger gene sets that may be derived from high-throughput experiments.
The next step is to choose the way the network is represented as features in the machine learning model.
For BioGIRD, using Adjacency or Embedding usually results very similar performance
For STRING-EXP, using Adjacency is usually the best feature representation.
For GIANT-TN, Influence is usually the best representation.
For STRING, if your input gene set size is smaller or if the gene set is similar to a specific biological process, then using Adjacency is the best choice. If the gene set size is larger or if the gene set corresponds to a complex phenotype, then use Influence.
The final step is to choose the background used to determine the genes used as negative examples in the machine learning model. If your gene set corresponds to a biological process or pathway, choose GO. Instead, if it corresponds more closely to a disease or a complex phenotype, then choose DisGeNet.
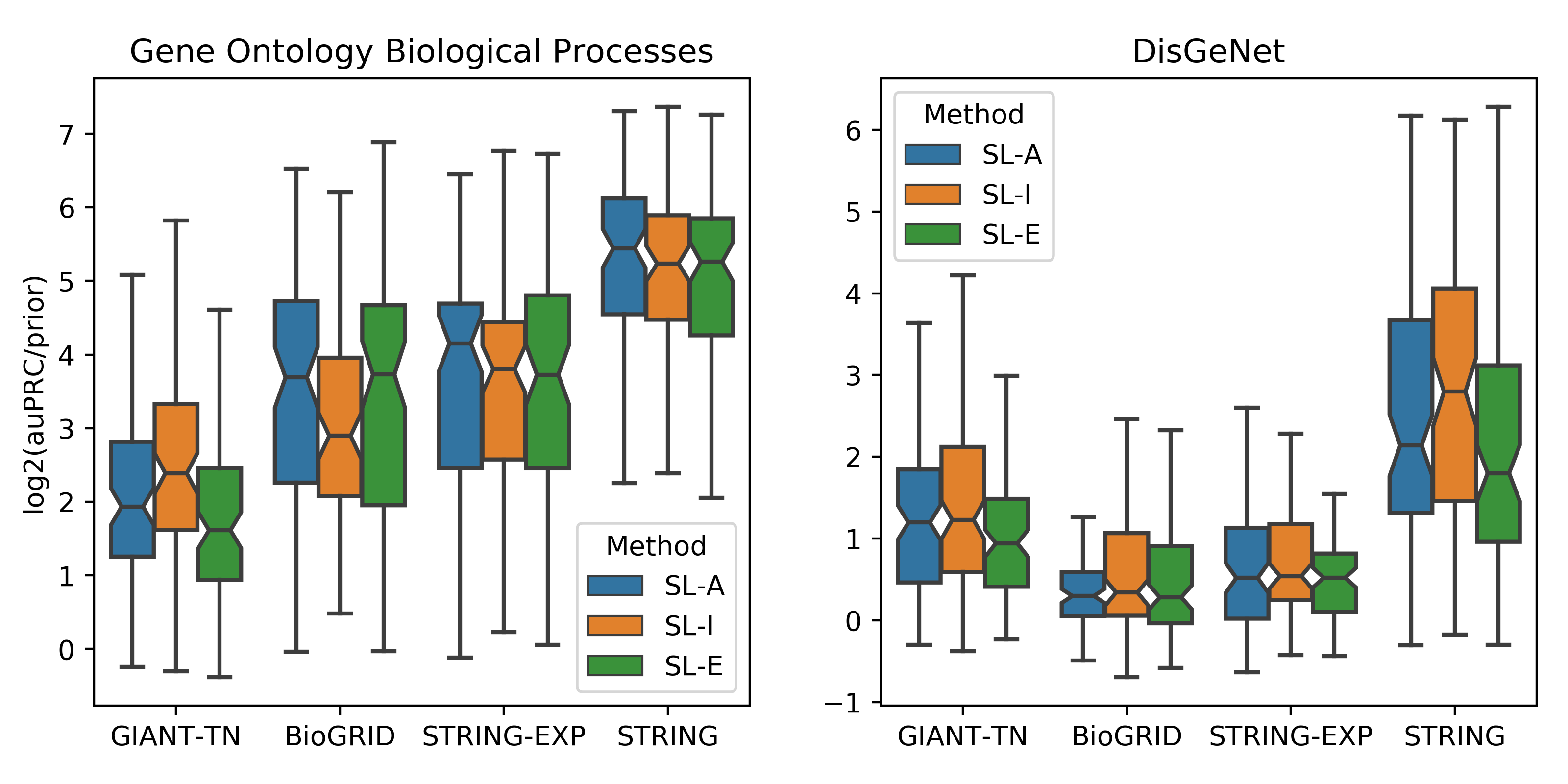
The above figure displays results from the [GenePlexus] paper Supervised learning is an accurate method for network-based gene classification where the parameters are options in the GenePlexus webserver. The left panel is for models trained for Gene Ontology biological prcoesses and the right panel is for models trained for DisGeNet diseases. Each boxplot contains the results of anywhere between 89 and 160 models. Each model was trained using the study-bias holdout method, where for each geneset in a geneset collection, the most well studied genes were used to train the model and the least studied genes were used for testing. This figure can be used to help users pick the network, feature type and negative selection class that best suits their input gene list. It is worth noting that the STRING network is built in part by using Gene Ontology and DisGeNet annotations, and this circularity could be the reason for the enhanced performance of the STRING network in this evaluation.