PyGenePlexus
PyGenePlexus is a Python package for running the [GenePlexus] model.
PyGenePlexus enables researchers to predict genes similar to an uploaded geneset of interest based on patterns of connectivity in genome-scale molecular interaction networks, with the ability to translate these findings across species.
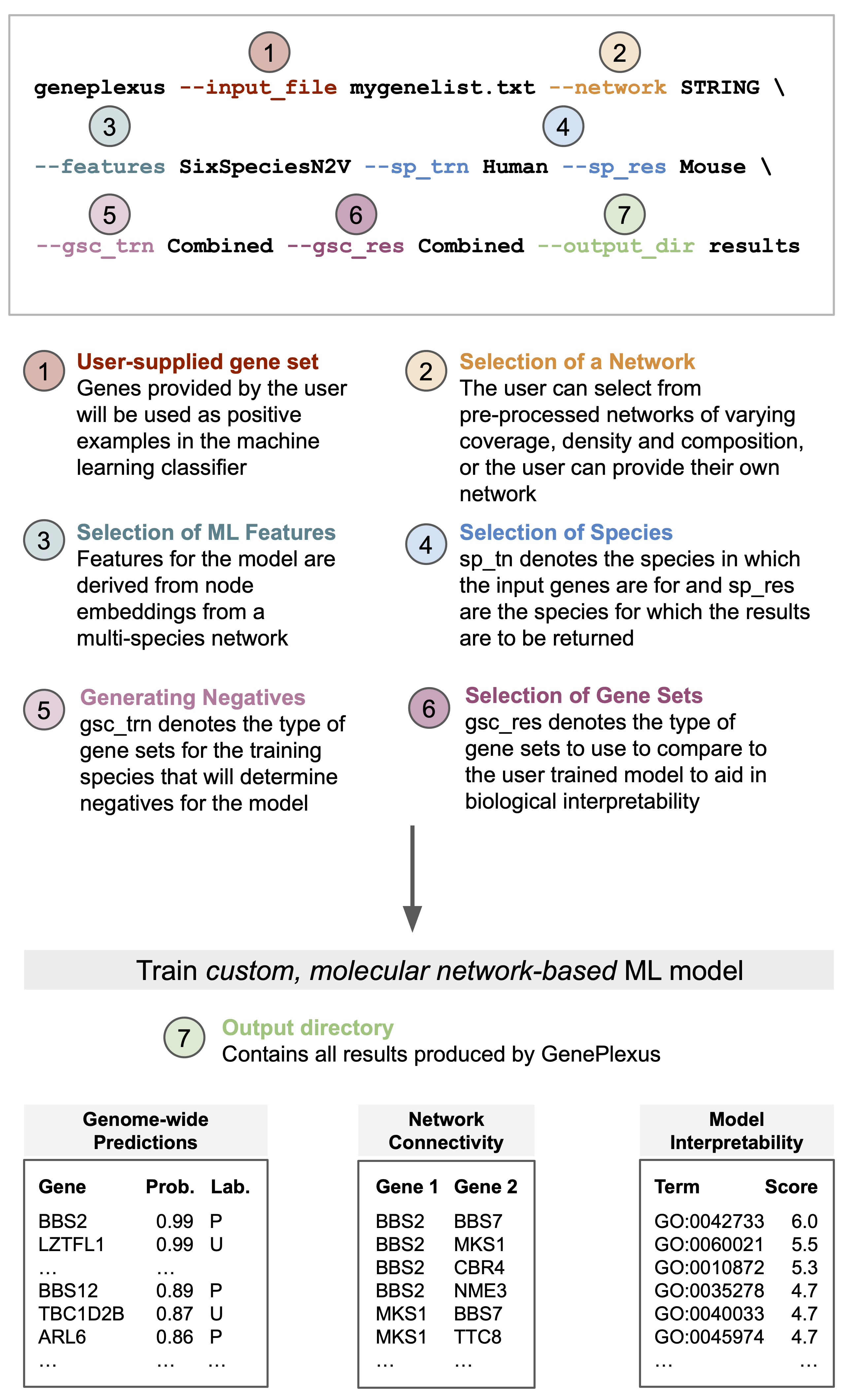
Overview of PyGenePlexus
Given a list of input genes and a geneset collection (GSC) to help select negative examples, the package trains a logistic regression model using node embeddings as features and generates the following outputs, either in the same species as the input genes or translated to a model species.
Genome-wide prediction of how functionally similar a gene is to the input gene list. Evaluation of the model is provided by performing k-fold cross validation. The default is 3-fold cross validation when a minimum of 15 input genes are supplied. These parameters can be changed when accessing the Python class. PyGenePlexus does not enforce a minimum or maximum number of genes, and we note evaluations of the model were carried out for gene sets ranging between 5 and 500 genes. See
fit_and_predict()(Optional) Interpretability of the model is provided by comparing the model trained on the user gene set to models pretrained on 1000’s of known gene sets from [GO] bioloigcal proceses, [Monarch] phenotypes and [Mondo] diseases. See
make_sim_dfs()(Optional) Interpretability of the top predicted genes is provided by returning their network connectivity.
make_small_edgelist()
Note
Links to other GenePlexus products
Quick start
PyGenePlexus comes with an easy to use command line interface (CLI) to run the full GenePlexus pipeline given an input gene list. Go get started, install via pip and run a quick example as follows.
pip install geneplexus
geneplexus --input_file my_gene_list.txt --output_dir my_result
Note that you need to supply the my_gene_list.txt file, which is a line
separated gene list text file (NCBI Entrez IDs, Symbol or Ensembl IDs are
accepted). An example can be found on the
GitHub page under
example/input_genes.txt. More info can be found in PyGenePlexus CLI.
Warning
All necessary files for a specific selection of parameters (network,
feature, species, and gene set collection) will be downloaded automatically and
saved under ~/.data/geneplexus. User can also specify the location of
data to be saved using the --output_dir argument. The example
provided will download files that occupy ~4GB of space.
Using the API
A quick example of generating predictions using an input gene list. More info can be found in PyGenePlexus API.
from geneplexus import GenePlexus
input_genes = ["ARL6", "BBS1", "BBS10", "BBS12", "BBS2", "BBS4",
"BBS5", "BBS7", "BBS9", "CCDC28B", "CEP290", "KIF7",
"MKKS", "MKS1", "TRIM32", "TTC8", "WDPCP"]
gp = GenePlexus(net_type="STRING", features="SixSpeciesN2V",
sp_trn="Human", sp_res="Human",
gsc_trn="Combined", gsc_res="Combined",
input_genes=input_genes, auto_download=True,
log_level="INFO")
df_probs = gp.fit_and_predict()[1]
print(df_probs.iloc[:10])
Note
v2 of PyGenePlexus is signifcanlty different than v1 and uses a different set of backend data, which only includes human data. For information of that version see https://pygeneplexus.readthedocs.io/en/v1.0.1/
Using PyGenePlexus
Package reference
Appendix